
Multiple-Sequence Alignment
Clustal
Clustal[1] has been part of the Sequencher family of plugins since version 4.9. It is a widely used multiple-sequence alignment program which works by determining all pairwise alignments on a set of sequences, then constructs a dendrogram grouping the sequences by approximate similarity and then finally performs the alignment using the dendogram as a guide. You can use Clustal to align your sequences directly from the Sequencher project window. Choose from a range of parameters to control the alignment process.
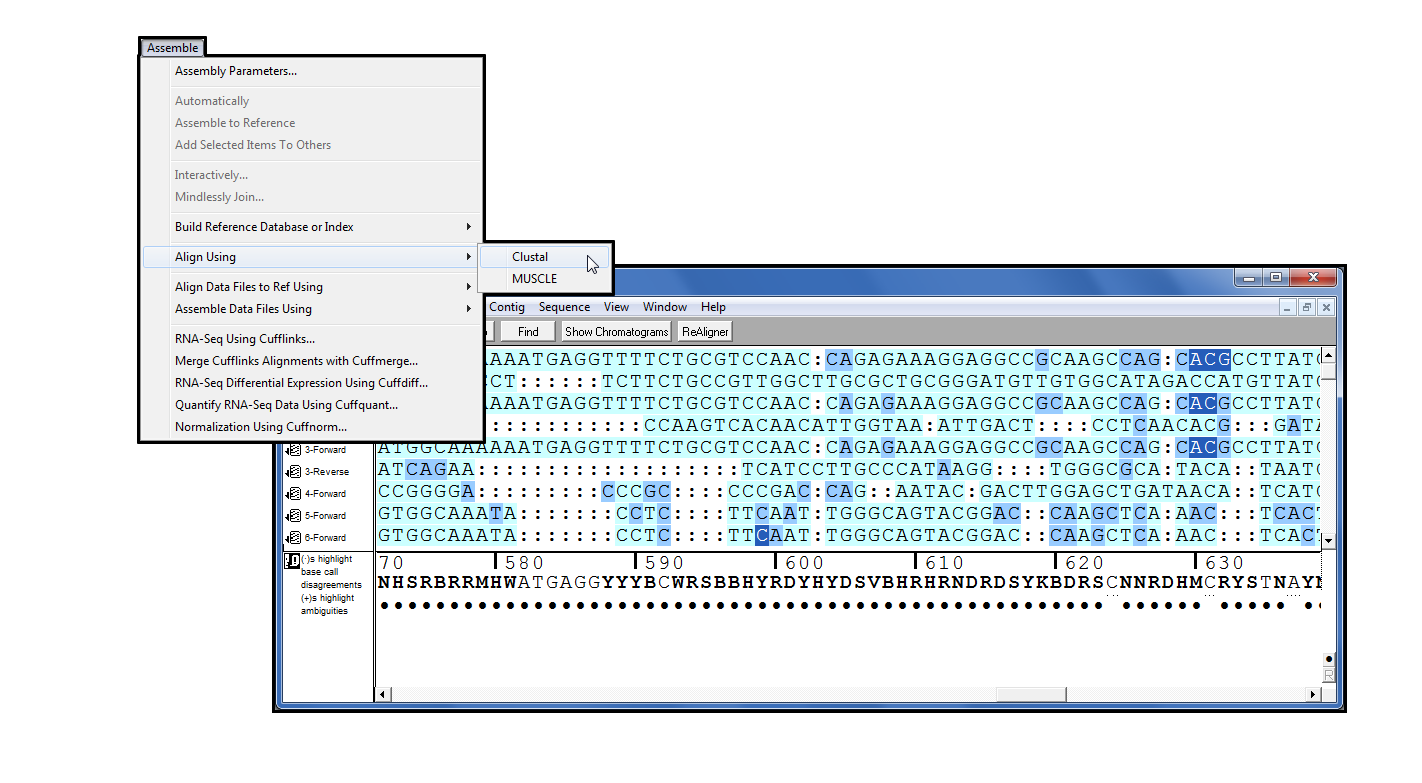
Once the alignment is complete you will see the results as a contig or contigs within Sequencher which you can subject to a variety of analyses.
Speed up your Clustal alignments by combining Clustal with the power of Sequencher's Assemble by Name functionality for aligning multiple sequences from different sources.
Finally, export the results in a variety of different formats such as MSF, Phylip, NEXUS and FastA for use in other programs or simply create a consensus and export it.
MUSCLE
MUSCLE[2], a multiple-sequence alignment (MSA) program, joins the Sequencher 5.1 family of plugins. It joins Clustal, making it the second MSA program in Sequencher’s DNA-Seq Tools. Boasting both speed and accuracy, it compares very favorably [3] to other multiple-sequence alignment programs.
MUSCLE is said to have four major steps in its alignment process. The first step constructs a distance matrix between pairs of sequences using k-mer clustering, this is then converted into a tree. The second step uses this tree to guide a progressive alignment. In the final two steps, the MUSCLE algorithm tries a number of different methods to see if it is possible to improve the tree and hence the multiple alignment.
Once the process is complete and MUSCLE has built the alignment, you will see the results as a contig within Sequencher. Use Sequencher’s tools to annotate your alignment or export your alignment and place it into a special phylogenetics program.
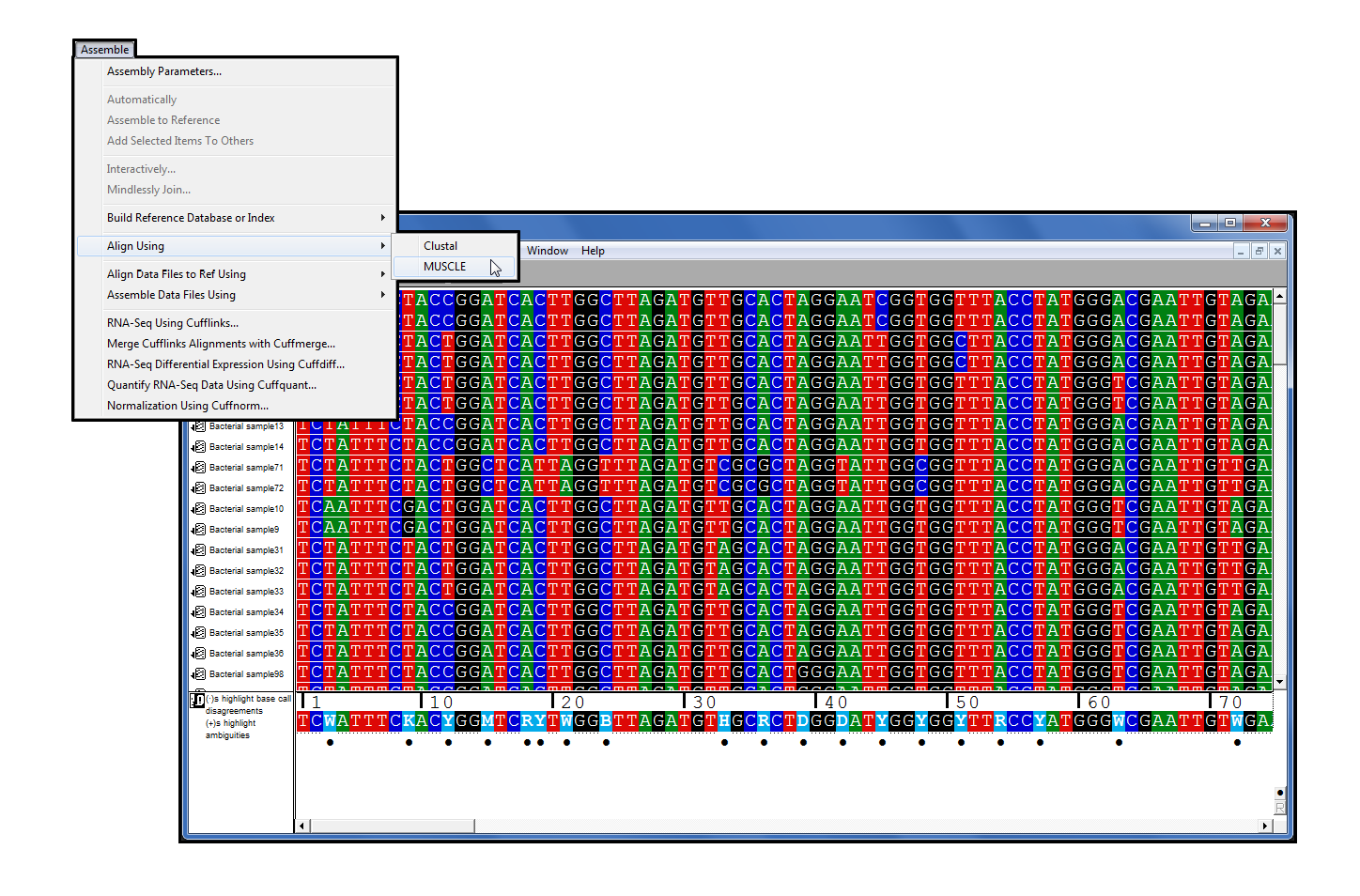
MUSCLE is a command line program, which means that normally you would be using this program through a Terminal application. Sequencher gives you access to MUSCLE’s power without the problems of learning to use the UNIX command line.
[1] Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD (2003). "Multiple sequence alignment with the Clustal series of programs". Nucleic Acids Res 31 (13): 3497-3500
[2] Nucleic Acids Res.2004 Mar 19;32(5):1792-7. MUSCLE: multiple sequence alignment with high accuracy and high throughput.Edgar RC.
[3] BMC Bioinformatics.2004 Aug 19;5:113. MUSCLE: a multiple sequence alignment method with reduced time and space complexity.Edgar RC.